萍乡市人民医院国家药物临床试验机构 临床试验安全性报告流程
萍乡市人民医院国家药物临床试验机构
临床试验安全性报告流程
一、个例安全性报告
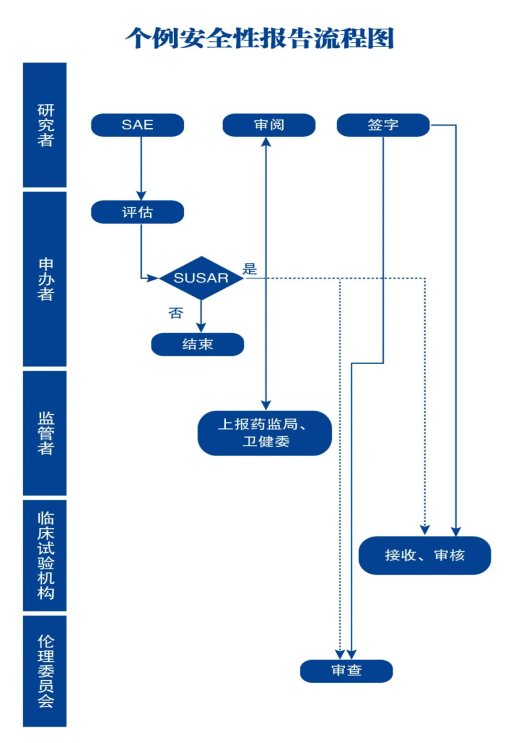
步骤1 研究者上报安全性事件
核心提示:研究者向申办者报告所有的SAE,并接收申办者评估完成的SUSAR报告,评估是否需要采取相关措施以保护受试者权益
(1)研究者应当立即向申办者书面报告所有SAE(研究者获知24h内),随后应当及时提供详尽、书面的随访报告。
(2)书面报告时应保证报告内容完整、准确,以提供申办者评估。
(3)相关性判断应当遵循方案规定,对“药物”-“事件”的相关性做出科学判断,并提供依据。
步骤2 申办者处理安全性事件
核心提示:申办者收到任何来源的安全性相关信息后,应当立即分析评估,基于事实作出科学独立的判断,包括严重性、与试验药物的相关性以及是否为预期事件等。
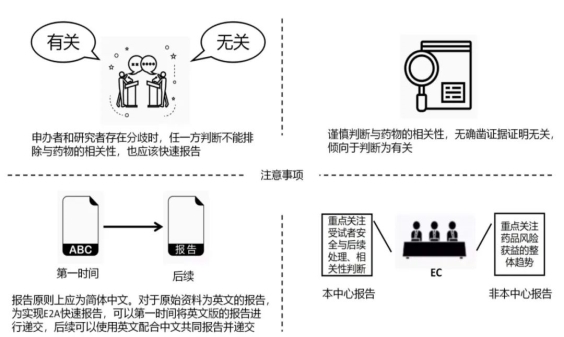
(1)不同意见的处理申办者在评估事件的严重性和相关性时,如果与研究者持有不同的意见,特别是对研究者的判断有降级的意见(如:将研究者判断为相关的事件判断为不相关),必须写明理由。在相关性判断中不能达成一致时,其中任一方判断不能排除与试验药物相关的,也应快速报告。
(2)保守原则 在撰写评估报告时,申办者需要明确相关性判断的依据。应谨慎地判断相关性,在无确凿依据判断无关时,倾向于判断为有关。
步骤3 SUSAR报告递交伦理、机构办
核心提示:申办者需要将SUSAR报告递交国家药监局、国家卫健委、研究者,研究者在收到申办方递交的SUSAR报告后,向所在伦理委员会、机构办递交SUSAR报告。
(1)关于报告时限的要求:
申办者应当将可疑且非预期严重不良反应快速报告给所有参加临床试验的研究者及临床试验机构、伦理委员会,参考时限如下:
①致死或危及生命的非预期严重不良反应,申办者应在首次获知后尽快报告,原则上不得超过7天,并在随后的8天内报告、完善随访信息。(申办者首次获知当天为第0天)。
②对于非致死或危及生命的非预期严重不良反应,申办者应在首次获知后尽快报告,原则上不得超过15天。
(2)关于报告文字的要求:报告需为简体中文,对于英文版的原始资料,第一时间可递交英文版资料,后期应尽快将中文翻译版进行补充递交,同时递交报告的汇总中文摘要或行列表。
(3)关于报告格式的要求:SUSAR报告的递交应采用标准化、结构化的信息,建议使用SAE/SUSAR报告表(见附件模板)或国际医学科学组织理事会(Councilfor international Organizations of Medical Sciences)表。
(4)关于报告方式的要求:以研究者签字递交的书面安全性信息报告为正式递交。递交过程可采取邮件(伦理邮箱:pxsrmyyec@qq.com、机构办邮箱:pxsrmyygcp@163.com)、研究者书面报告递交两种方式相结合。
二、研发期间安全性更新报告(DSUR)
研发期间安全性更新报告(Development Safety Update Report,以下统称:DSUR)的主要目的是对报告周期内收集到的与在研药物(无论上市与否)相关的安全性信息进行全面深入的年度回顾和评估。
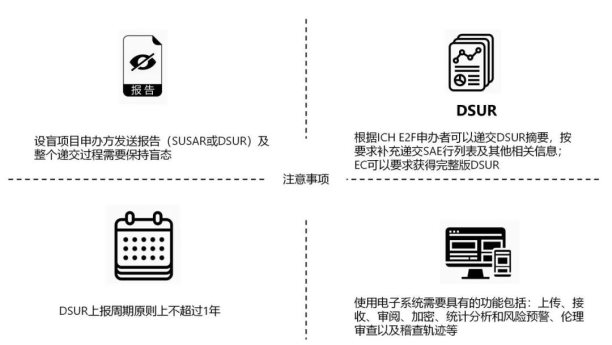
(1)关于报告时限的要求:研发期间安全性更新报告进行年度报告递交,原则上报告周期不超过一年。
(2)关于报告文字的要求:报告需为简体中文,对于英文版的原始资料,需中文翻译版和英文版一并递交,同时递交汇总的中文摘要或行列表。
(3)关于报告格式的要求:
按照ICH E2F的建议,申办者可以使用DSUR的执行概要进行递交,并按相应要求补充递交严重不良反应行列表,简要说明DSUR报告周期内,获得的有效性和安全性信息的任何变更及已经采取或将要采取的解决临床研发项目中新出现的安全性问题的措施。
DSUR的具体撰写与递交要求,请参照ICH-E2F及CDE和即将发布的《研发期间安全性更新报告要求及管理规定》。具体但不限于以下:
①前言,包括报告周期和报告序列号
②研究药物 -作用机理、治疗分类、适应症、剂量、给药途径、剂型
③研究的适应症和人群
④临床试验的涵盖范围
⑤简要说明并解释 DSUR中未包含的信息,对单个研究药物递交多个 DSUR的理由(如适用)
⑥受试者累计暴露的预估
⑦上市状态(全球范围内)
⑧整体安全性评估的摘要
⑨重要风险的摘要
⑩包括研究者手册重大改动在内的出于安全性考虑而采取的行动
⑪结论,风险获益相关的结论
(4)关于报告方式的要求:
按照2020版GCP要求,作为阶段性的安全性汇总,申办者需要将DSUR报告递交国家药监局、国家卫健委、研究者;研究者收到申办者提供的DSUR纸质报告后应当及时签收审阅,并向机构办、伦理委员会递交。
以研究者签字递交的书面安全性信息报告为正式递交。递交过程可采取邮件(伦理邮箱:pxsrmyyec@qq.com、机构办邮箱:pxsrmyygcp@163.com)、研究者书面报告递交两种方式相结合。
附件:SUSAR/严重不良事件报告表(本模板供参考,请按照新版GCP(2020年)要求,并结合申办者安全性信息要素内容进行撰写)
(完)
参考法规:
1.《药物临床试验质量管理规范》,2020,国家药品监督管理局、国家卫生健康委员会
2. ICH E6(R2)Good Clinical Practice,2016,ICH
ICH《E2B(R2)安全性消息处理和个例安全性报告技术规范》,2018,国家药品监督管理局
《药物临床试验期间安全性数据快速报告标准和程序》,2018,国家药品监督管理局
ICH E2F Development safety updatereport,2011,ICH
ICH E2A Clinical Safety DataManagement:Definitions and Expedited Reporting, 1994, ICH
《临床试验安全性报告工作指引》,2020,中国临床研究能力提升与受试者保护”高峰论坛(CCHRPP)
附件: SUSAR/严重不良事件报告表
新药临床研究批准文号: | 中心号: | 报告类型:□首次 □随访 □总结报告 |
申办方临床研究方案号: | 受试者编号: | 报告编号: |
研究项目及报告单位信息 | 报告时间 | 年 月 日 | |
医疗机构及专业名称 | 电话 | ||
申报单位名称 | 电话 | ||
研究方案名称 | 临床试验适应症 | ||
临床研究分类 | □Ⅰ期 □Ⅱ期 □Ⅲ期 □Ⅳ期 □生物等效性试验 □临床验证 | ||
报告者信息 | 获知时间 | 年 月 日 | |||
报告姓名 | 报告者职业 | 电话 | |||
报告者地址 | 邮箱 | ||||
患者信息 | |||||||||
姓名缩写 | 出生日期 | 性别 | □男□女 | 身高(cm) | 体重(kg) | ||||
受试者编号 | 民族 | 发生SAE时年龄 | 受试者是否退出研究 | □是 □否 | |||||
患者死亡 | □是□否 | 死亡时间 | 死亡原因 | 是否尸检 | □是□否 | 尸检结果 | |||
相关病史与治疗 | □不详 □无 □见下表 | ||||
现病史 | 试验用药适应症以外,SAE发生时未恢复的疾病 | ||||
疾病名称 | 开始时间 | 是否持续 | 结束时间 | 治疗药物通用名称 | 用法用量 |
既往病史 | 试验用药适应症以外,SAE发生时已经恢复的疾病 | ||||
疾病名称 | 开始时间 | 是否持续 | 结束时间 | 治疗药物通用名称 | 用法用量 |
饮酒史 | □无 □有 | 吸烟史 | □无 □有 | 家族史 | □无 □有 |
肝病史 | □无 □有 | 胃病史 | □无 □有 | 过敏史 | □无 □有 |
与SAE相关实验室检查项 | □不详 □无 □见下表 | ||||
检查名称 | 检查日期 | 检查结果 | 正常值上限 | 正常值下限 | 备注 |
合并用药 □不详 □无 □见下表 注:合并用药是指SAE发生前开始使用,SAE发生时正在使用的药品;针对SAE的治疗用药,请填写在“SAE发生及处理的详细情况”栏。 | |||||||
药物名称 | 使用原因 | 剂量/剂量单位 | 剂型 | 频次 | 给药途径 | 开始时间 | 结束时间 |
试验用药使用情况(如有多个试验用药,请复制此表格添加) (若有除试验药物外的怀疑药品及相互作用的药物,请复制并添加此表格;如果是盲态试验请填写研究药品名称/安慰剂或对照药) | |||||||
试验用药品中文名称 | 研究设计 | ||||||
试验用药品英文名称 | 用药原因 | ||||||
是否已给药 | □是 □否 | 药物编号 | |||||
是否已破盲 | □是 □否,破盲日期: | 破盲原因 | |||||
对试验药物采取的措施 | □继续用药 □减小剂量 □停用药物 □停用药物又恢复 □不适用 □不详 □增加剂量 | 采取措施时间 | |||||
剂量详情 | |||||||
剂量/计量单位 | 给药途径 | 频次 | 剂型 | 开始日期 | 结束日期 | ||
严重不良事件(此表可复制) | ||||
SAE1 | SAE2 | SAE3 | SAE4 | |
不良事件名称(诊断) | ||||
开始日期 | ||||
结束日期 | ||||
研究者获知SAE时间 | ||||
严重性标准 | □导致死亡 □致残/致功能丧失 □危及生命 □导致住院或延长住院时间 □致畸/致出生缺陷 □其他重要医学事件 | □导致死亡 □致残/致功能丧失 □危及生命 □导致住院或延长住院时间 □致畸/致出生缺陷 □其他重要医学事件 | □导致死亡 □致残/致功能丧失 □危及生命 □导致住院或延长住院时间 □致畸/致出生缺陷 □其他重要医学事件 | □导致死亡 □致残/致功能丧失 □危及生命 □导致住院或延长住院时间 □致畸/致出生缺陷 □其他重要医学事件 |
严重程度 | □轻度 □中度 □重度 | □轻度 □中度 □重度 | □轻度 □中度 □重度 | □轻度 □中度 □重度 |
CTCAE分级 | ||||
国内SAE报道情况 | □有 □无 □不详 | □有 □无 □不详 | □有 □无 □不详 | □有 □无 □不详 |
国外SAE报道情况 | □有 □无 □不详 | □有 □无 □不详 | □有 □无 □不详 | □有 □无 □不详 |
不良事件结果 | □不详 □死亡 □未好转 □好转 □痊愈 □痊愈伴有后遗症 | □不详 □死亡 □未好转 □好转 □痊愈 □痊愈伴有后遗症 | □不详 □死亡 □未好转 □好转 □痊愈 □痊愈伴有后遗症 | □不详 □死亡 □未好转 □好转 □痊愈 □痊愈伴有后遗症 |
是否针对SAE进行治疗 | □不详 □无 □是,需要事件描述说明 | □不详 □无 □是,需要事件描述说明 | □不详 □无 □是,需要事件描述说明 | □不详 □无 □是,需要事件描述说明 |
相关性评价 (不良事件—怀疑药物) 研究/怀疑药物名称1: | □肯定有关 □很可能有关 □可能有关 □可能无关 □肯定无关 □无法评价 | □肯定有关 □很可能有关 □可能有关 □可能无关 □肯定无关 □无法评价 | □肯定有关 □很可能有关 □可能有关 □可能无关 □肯定无关 □无法评价 | □肯定有关 □很可能有关 □可能有关 □可能无关 □肯定无关 □无法评价 |
停用研究/怀疑药品1后SAE是否消失? | □是 □否 □不详 □不适用 | □是 □否 □不详 □不适用 | □是 □否 □不详 □不适用 | □是 □否 □不详 □不适用 |
再次使用研究/怀疑药品1后,事件是否再次出现? | □是 □否 □不详 □不适用 | □是 □否 □不详 □不适用 | □是 □否 □不详 □不适用 | □是 □否 □不详 □不适用 |
SAE发生及处理的详细情况 | |
研究者签名: | 日期: |

